Classified as uterine or nonuterine
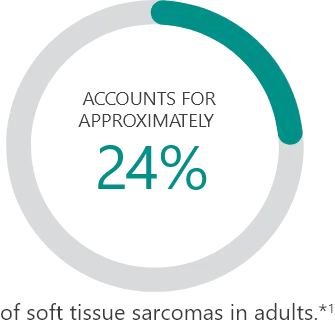
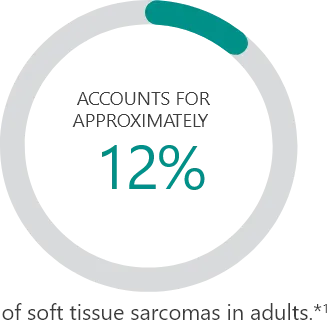
Subtypes2:
Percentages based on incidence data from 1978-2001 collected in the 9 Surveillance, Epidemiology and End Results (SEER) program areas. Cases of soft tissue sarcoma were categorized into major histologic types and subtypes, according to criteria specified in the 2002 World Health Organization Classification of Tumors and the recommendations of an expert pathologist. Additional cases that met the morphologic criteria of soft tissue sarcoma regardless of primary site, except bones and joints, were added for a total of 26,758 cases used in the analysis.
Chemosensitivity of histological subtype3
Natural history3
Tumor burden3
Tumor stage4
Tumor grade4
Patient’s general condition and level of symptoms3
Comorbidities3
Previous treatments3
Patient goals3
PFS allows new treatments to be evaluated in smaller studies and over shorter periods of time, and results are not confounded by subsequent lines of therapy.5
Explore a treatment option for liposarcoma and leiomyosarcoma.

YONDELIS® is the only treatment, after an anthracycline-containing regimen, approved specifically for both:
REFERENCES:
